醫療器械淨化解決方案(àn)及規範
1、GB50073-2013潔淨廠(chǎng)房設計規範
2、醫(yī)藥工(gōng)業潔淨廠房設(shè)計規範(fàn) GB 50457-2008
3、體外(wài)診斷試劑(jì)生產實施細則(試行)
4、YY/T 1244-2014 體外診斷試劑用純化水
5、中國藥典GMP2015版(bǎn)純化水(shuǐ)標準
6、《潔淨室施工(gōng)及驗收規範》GB50591-2010
7、無菌醫療器具生產管理規範 - YY 0033-2000
醫療器械GMP車間解決方案:
1、在生產或使用中活性物質、滅活物質的汙染(包括熱原)對產品產生重要影響的植入性醫療器(qì)械,醫(yī)療(liáo)器械GMP車間規劃設(shè)計,應對工作環境進行控製,對滅活的方法應予驗證並保(bǎo)存(cún)記錄。此類產品的生產和包裝應在有規範要求的、可控的環境下進行。
2、對非無菌植入性醫療器械或使用前預期(qī)滅菌的醫(yī)療器(qì)械,如果通過確(què)認的產品清潔、包裝過程(chéng),能將汙染降低並保持一致的控製(zhì)水平(píng),醫療器械GMP車間施工,應建立一個(gè)受控的環境來包含(hán)該確認的清潔和包裝(zhuāng)過(guò)程。生產企業可參照YY0033-2000標準或自行驗證並確定產品的生產潔淨級別。
3、應對(duì)受汙染或易於汙染的產品進行控製。應對受汙染或易於汙染(rǎn)的產(chǎn)品、工(gōng)作台麵或人員建立搬運(yùn)、清潔和除汙染的文件。
醫療器械GMP車間施工設計參考:
1、《醫療器械生產企業質量(liàng)管理規範(試行)》,國家食品藥品監督管理(lǐ)局(jú)(2009年)--2015廢止。
2、《體外診斷試劑生產實施(shī)細則(zé)(試行)》,國家食品(pǐn)藥品監(jiān)督管理局(2007年)--2015廢止。
3、《關於實施(醫療器械生產質量管理規(guī)範(試行))及其配套文件有關問題的通知》(2011年)--2015廢止。
前麵1~3的2007、2009年(nián)醫療器械的規範、細則、標準在2015年停用,醫療器械GMP車間規劃,代之2015的醫療器械生產企業質量管理規範及無菌、植入、體(tǐ)外診斷試劑三個附錄。
4、醫藥(yào)工業潔(jié)淨廠房設計規範 GB 50457-2008
5、體外診斷試劑生產實施細則(試行)
6、YY/T 1244-2014 體外診斷試劑用純(chún)化(huà)水
7、中國藥典GMP2015版純(chún)化水標準
8、《潔淨室施工及驗收規範》GB50591-2010
9、無菌醫療器(qì)具生產管理規範 - YY 0033-2000
10、《潔淨廠房設(shè)計規範》(GB50073-2010)
11、《潔淨室施工及(jí)驗收規範(fàn)》(GB50591-2013)
12、《醫療產品的無菌(jun1)加工第1部分:通用要求》(YY/T0567.1-2005)
13、《無菌醫療器械生產與質量管理講義(yì)》,國家藥品監(jiān)督管理局(2000)
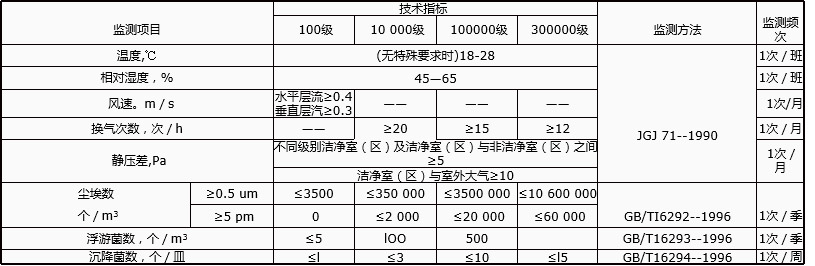
為確認A級潔淨區的級別,每個采(cǎi)樣點的采樣量不得少於1立方米。A級潔淨區空氣懸浮粒子的級別為ISO4.8,以≥5.0μm的懸浮粒子為(wéi)限度(dù)標準(zhǔn)。B級潔淨(jìng)區(靜(jìng)態)的空氣懸浮粒子(zǐ)的級別為ISO5,同時包括表(biǎo)中兩種粒徑的懸浮粒子。對於C級潔淨區(qū)(靜態和動態)而言(yán),空氣懸浮粒子的級(jí)別分別為ISO7和ISO8。對於(yú)D級潔淨區(靜態)空氣懸浮粒(lì)子的級別為ISO8。測試方法可(kě)參照ISO14644-1。
(2)在確(què)認級別(bié)時,應當使用(yòng)采(cǎi)樣管較短的便攜式塵埃粒子計數器(qì),避免(miǎn)≥5.0μm懸(xuán)浮粒子在遠程采樣(yàng)係(xì)統的(de)長采樣管中沉降。在單向流係統中,應當采用等動力學的(de)取樣頭。
(3)動態測試可在常規操作、培養基模擬灌(guàn)裝過程中進行,證明達到動態的潔淨度級別,但培養基模擬灌裝試驗要求在“較差狀況”下進行動態測試。
根據相關規範要求,對無菌醫療器械生產車間、藥品生產車間、醫學生(shēng)物學實驗(yàn)室、手術室等都(dōu)要求建設符合相(xiàng)關標準的潔淨室。在潔淨室建設或改建時,不能依賴於較終的竣工驗收來保證潔淨室(shì)的(de)質量,必須從設計及設備選型階段就嚴格把關,在建設的全過程中對主(zhǔ)要關鍵點嚴格檢查、監督,在實際使用中(zhōng)定期監測才能保證(zhèng)潔淨室達(dá)到設計指標和使用要求。
無菌醫療器械是任何標明(míng)“無菌”的醫療器械,生產潔淨室是保證無菌醫(yī)療器械質量的基本條件,控製(zhì)無菌醫療器械生產過程的環境並規範其生產,防止環境對無菌醫療器械汙染,潔淨室必須滿足規定環境參數的要求來建設和定期監測。
醫療器械(xiè)淨化工程建設中(zhōng)需考慮從以下(xià)問題:
1.醫療器械包裝車間潔淨室工程所需要的(de)淨化材料;
2.醫療器械廠房潔淨室及醫療器械(xiè)包裝(zhuāng)車間潔淨室(shì)工程的設計、安裝、調試、維護等綜合服務;
3.醫療器械包裝車間潔淨室工程空調淨化部分。
溫度和相對(duì)濕度
無菌醫療器械在無特殊規定時,通常要求溫度在法規標(biāo)準檢測StandardandTesting18~28C,濕度在45%~65%,企業一般都可以控(kòng)製在要求內。如(rú)在動態監測中發現達不到要求,可能是(shì)室內有(yǒu)產熱大的儀器設備。
風量、換氣次數、靜壓差
在潔淨室體積確定的情況下,換氣次數由該室的送風量決定,而靜壓差取決於房間的送風量與回風量、排風量的差值。係統總送風量(liàng)、新風量、總排風量和對外壓差可以通過調整風機頻率轉速或總閥門開啟度來實現,各房間(jiān)的風量和壓力則可通過調整分支管路(lù)閥(fá)門(mén)開度來現(xiàn)。
懸浮粒子(zǐ)、浮(fú)遊菌、沉降菌
測試條件如不能滿足規定的環(huán)境參數(溫濕度、風速(sù)、換氣次(cì)數、靜(jìng)壓(yā)差在規定範圍之內)要求,關鍵項目懸浮粒子、浮遊菌(jun1)或沉降(jiàng)菌(jun1)的測試結果應視為無效。由於(yú)溫度、相(xiàng)對濕度、風(fēng)速、換氣次數、靜壓差共同構成了潔淨室的微氣候,是潔淨室維護正常與(yǔ)否的重要(yào)指征,可(kě)將關(guān)鍵工序關鍵項目測試修(xiū)訂為關鍵工序全(quán)性能(néng)測試。隻(zhī)有這樣,才能全麵、係(xì)統監測生(shēng)產潔淨室,為確保潔淨室性能監測的數據科學(xué)性、準確性,測試部門在進行關(guān)鍵(jiàn)項目懸浮粒子、微生物測試時,應同時進行溫度、相(xiàng)對濕度、換氣次數、靜壓(yā)差等前(qián)提條件的(de)測試。
溫度(dù)
潔淨室夏季(jì)室溫超過設計範圍的原因,多是由(yóu)於開始確定的各潔淨室的空調送風量(liàng)即換氣次數時隻注重滿足潔淨度指標(biāo),忽視了對各潔淨室熱平衡的校核計算。因此在生產(chǎn)潔淨室(shì)的設計(jì)及運行過程(chéng)中,必須對潔淨室的空調送風參(cān)數進行(háng)實(shí)時修正,保證(zhèng)各個季節生產潔淨室的(de)溫(wēn)度都維持(chí)18~28C。溫度和(hé)相對濕(shī)度主要(yào)影響產品生產(chǎn)工藝及細菌的繁殖條件,還能引(yǐn)發由生產操作(zuò)人員舒適度對產品質量(liàng)的影響。
送風量、換氣次數
醫療器械淨化工程-無菌潔淨室工程設計階段(duàn)對送風量的確定,首先要滿足相應(yīng)潔淨度級別的(de)換氣次數要求,同時還要通過熱、濕負(fù)荷校核來(lái)進一步確定風量,在此基礎上對(duì)高效過濾器進行選用。過濾器的處理風量應小於或等於額定風量(liàng),設置在(zài)同(tóng)一潔淨區(qū)內的高效(亞高(gāo)效、超高(gāo)效)空氣過濾器的阻(zǔ)力、效率(lǜ)宜接近。
醫(yī)療器械GMP車間管控(kòng)總要(yào)求:
(1)表麵平滑;(2)表(biǎo)麵有耐磨性;(3)良好的熱絕緣性;(4)不易產生靜電;(5)不吸濕,不透濕;(6)吸聲性好;(7)容易加工;(8)表麵不易附著灰塵;(9)容易除(chú)去附著(zhe)的(de)灰塵;
無菌醫療器具潔淨室(區)環境要求及監鍘
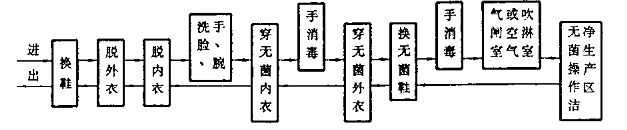
人員進出潔(jié)淨生產(chǎn)區的一般程序
人員進出潔淨生產區和(hé)無菌操作潔淨生(shēng)產區的一般程序(xù)見(jiàn)圖D1和圖D2。
圖D1 人員進出潔淨生產區的程序圖例
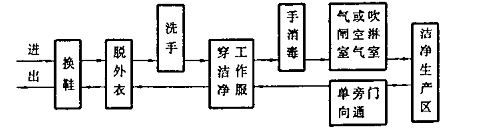
圖D2人員進出無菌操(cāo)作潔淨生產(chǎn)區的程序圖例